|
|
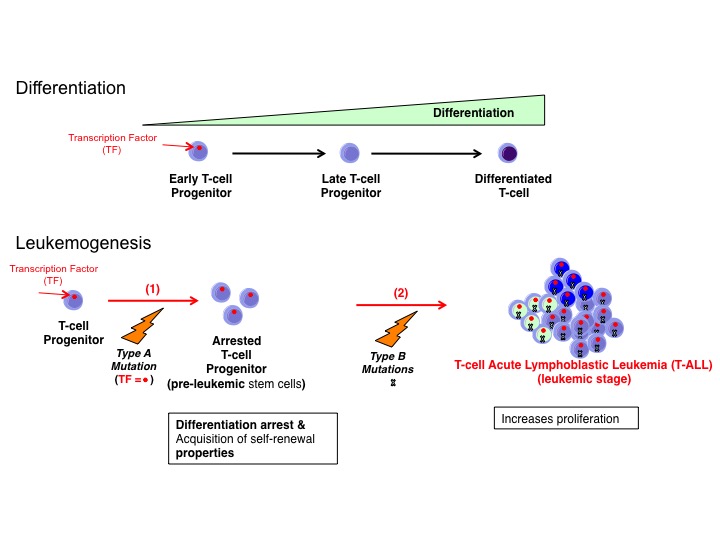
2- Understanding the molecular mechanism driving the development and maintenance of T-cell acute lymphoblastic leukemia (T-ALL) T-cell Acute Lymphoblastic Leukemias (T-ALLs) are aggressive hematological tumors that result from the malignant transformation of immature thymocytes. T-ALL is induced by genetic lesions that result from aberrant genomic rearrangements that occur during normal T lymphocyte development (“Type A mutations”). These rearrangements can give rise to the formation of oncogenic fusion proteins, but most often they bring proto-oncogenic transcription factors (TFs) under the transcriptional control of strong T cell specific enhancers. Indeed, 70% of genetic lesions in T-ALL result in the aberrant activation (i.e. misexpression) of oncogenic TFs that otherwise act as master regulators of other blood lineages during normal hematopoiesis. Oncogenic TFs are responsible for driving T cell progenitors to a pre-leukemic stage through an arrest in differentiation and the acquisition of self-renewal properties. Secondary mutations (“Type B mutations”) are then required for inducing full-blown leukemia through increased survival and proliferation. Our goal here is to understand the mechanism of leukemia development and maintenance by oncogenic TFs in T-ALL such that novel targeted therapeutic approaches that are more efficient and less toxic can be developed.
Focusing on the transcription factor TAL1 that acts a major oncogene in the T cell lineage whereas it controls normal development of other hemato-endothelial lineages, we performed a comparative analysis of TAL1 genome-wide binding in erythroid cells vs. T cell acute lymphoblastic leukemia (T-ALL). This study identified, for the first time, the leukemic gene network controlled by TAL1 in T-ALL (EMBO J 30: 494-509, 2011). Furthermore, we made the unexpected observation that the transcription factor RUNX1 plays a key role in guiding TAL1 to aberrant genomic loci for modifying expression of genes involved in leukemia maintenance. This was the first evidence implicating RUNX1 as an important player in T-ALL.
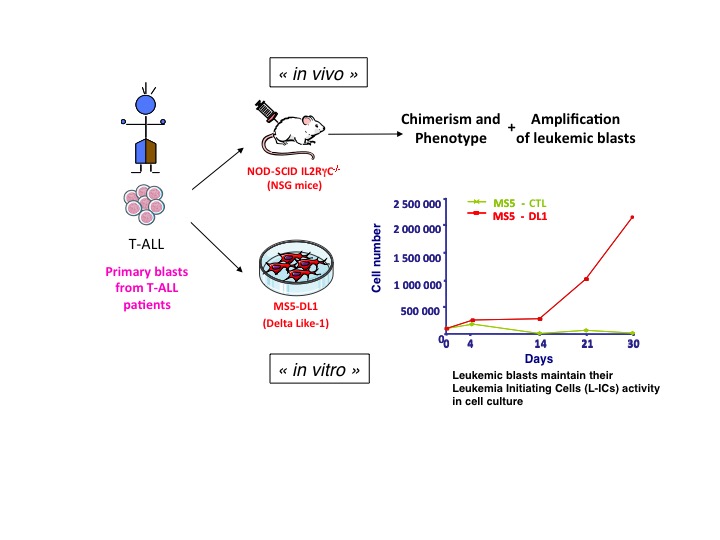
Currently, we are pursuing our studies to understand the molecular mechanisms underlying the TF-based epigenetic regulation of leukemia in T-ALL using complementary in vitro and in vivo approaches including patients-derived xenotransplantation models of leukemia. We expect that these studies will provide us with novel drug targets to eliminate human leukemic blasts, paving the way for personalized medicine in T-ALL.
|