|
|
1- Understand the molecular mechanism that controls erythropoiesis and its deregulation in ß-thalassemia The production of functional hemoglobin-expressing red blood cells from hematopoietic stem cells (HSCs) is a multi-step process guided by stage-specific transcription factors (TFs). These TFs control the expression of genes that drive erythropoiesis while simultaneously counteracting alternative cell fates, and they also ensure normal expression of ß-globin. Genetic defects that lead to decreased transcription of the adult ß-globin gene are at the origin of the hemoglobin disorder ß-thalassemia.
Our goal here is to decipher the transcriptional regulatory network that controls erythropoiesis in health and disease.
Our approach involves the isolation of endogenous TFs at various stages of erythroid differentiation and identifying their interacting partners by mass spectrometry. Notably, we are using Quantitative Proteomics (iTRAQ) methods to pinpoint the dynamics of protein interactions within the transcriptional regulatory network (Methods Mol Biol. 359:17-35, 2007). We expect that understanding the dynamic changes in transcription factors’ interactions during cell differentiation will allow us to better control cell fate decisions. For example, we have shown that the bZIP protein MafK regulates ß-globin expression by exchanging its heterodimerization partner from the repressor Bach1 to the activator p45 during erythroid differentiation (Nature Structural and Molecular Biology, 11 (1): 73-80, 2004). We are also using chromatin immunoprecipitation to study targeting of the identified transcription factors and cofactors to specific genes and/or genome wide as well as the resulting epigenetics modifications (The EMBO Journal, 30 (3): 494-509, 2011).
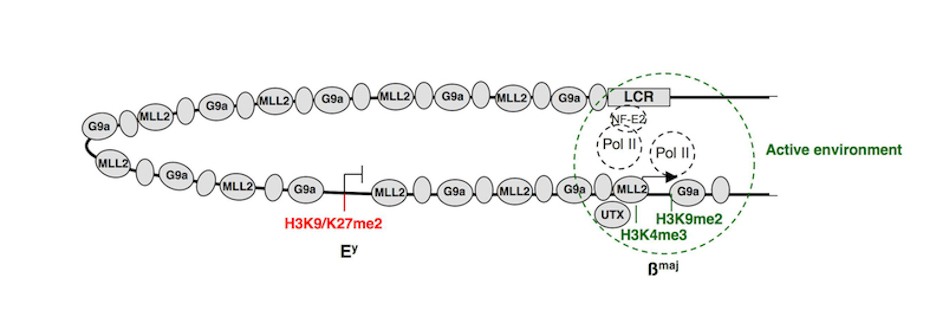
The ß-globin locus is one of our major model systems in erythropoiesis. For example, we have shown that during erythroid differentiation, the hematopoietic activator NF-E2 is implicated in recruiting the H3K4 methyltransferase complex MLL2 to the ß-globin locus. Following its recruitment to a distal DNA regulatory region, MLL2 is transferred to the active ß-globin gene over a distance of 40 kb via a “spreading” mechanism (Molecular Cell, 27: 573-584, 2007). Focusing on the H3K9 histone methyltransferase G9a/KMT1C, we showed that this enzyme is involved in maintaining the embryonic ß-globin gene in a repressed state while simultaneously activating the adult ß-globin genes in adult erythroid cells. Importantly, we demonstrated that the dual function of G9a as a coactivator vs. a corepressor entails its association within two distinct protein complexes, one containing the co-activator Mediator and one containing the co-repressor Jarid1a/KDM5A. Functionally, the repressive role of G9a relies on both methylation of histones H3K9/K27 and a coordinate action with the histone H3 lysine 4 (H3K4) demethylase Jarid1a for the maintenance of gene repression. In contrast, the activating role of G9a is independent of its methyltransferase activity and involves stabilization of the Mediator complex and cooperation with the H3K27 demethylase UTX. (PNAS, 106 (43): 18303-18308, 2009) (PNAS, 109 (46): 18845-18859, 2012) & (Epigenetics 5 (4), 2010).
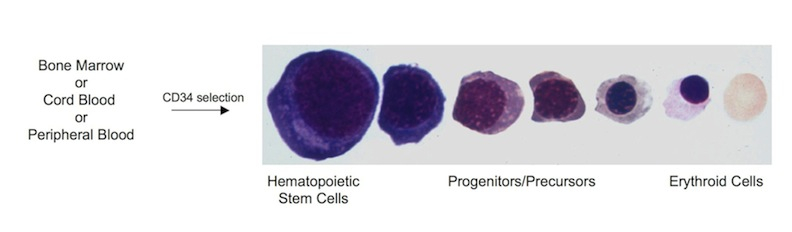
Currently, we are using a systems biology approach to build a network model of erythropoiesis based on dynamic changes in TF protein levels during erythroid differentiation of human hematopoietic stem/progenitor cells. Using a combination of relative (iTRAQ) and absolute (MRM) quantitative proteomics approaches, together with genomic approaches (RNAseq) in an ex vivo erythropoiesis differentiation system that we have developed (Journal of Visualized Experiments doi: 10.3791/2813), we aim to identify and quantify the dynamic changes in the ensemble of TFs and cofactors that occur during erythroid differentiation. Mathematical modeling will then allow us to integrate the dynamic and quantitative nature of the proteome into the transcriptional network of erythropoiesis. This improved network model is expected to serve as the benchmark for healthy erythropoiesis against which to compare erythroid-related disease states. This project is performed in collaboration with Jeff Ranish (ISB, Seattle, WA) and Ted Perkins (OHRI, Ottawa, ON).
|